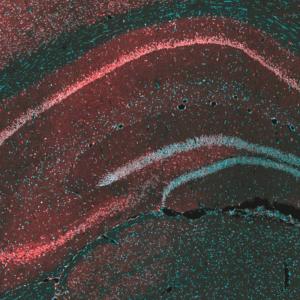
Researchers disabled memory formation in mice by removing the shuttle protein gamma-CaMKII. Selective replacement of the missing protein in nerve cells of a key brain region—the hippocampus, labeled green—resulted in full recovery of the ability to memorize.
Photo: NYU Langone Staff
Unable to carry signals based on sights and sounds to the genes that record memories, a broken shuttle protein may hinder learning in patients with intellectual disability, schizophrenia, and autism.
This is the implication of a study led by researchers at NYU School of Medicine and published online June 22 in Nature Communications.
Specifically, the research team found that mice genetically engineered to lack the gene for the gamma-CaMKII shuttle protein took twice as long as normal mice to form a memory needed to complete a simple task.
“Our study shows for the first time that gamma-CaMKII plays a critical role in learning and memory in live animals,” says Richard Tsien, PhD, chair of the Department of Neuroscience and Physiology and director of the Neuroscience Institute at NYU Langone Health.
“Adding more weight to our results, we showed that making the same change in the shuttle’s structure seen in a human child with severe intellectual disability also took away the ability of mice to learn,” says Dr. Tsien, also the Druckenmiller Professor of Neuroscience. He says this result suggests that the shuttle works similarly in the two species.
The research team then restored the learning ability by re-inserting the human version of the shuttle protein into mice.
The current study revolves around the nerve cells that coordinate thought and memory. Each cell in a nerve pathway sends an electric pulse down its branches until it reaches a synapse, a gap between itself and the next cell in line. Signals that form memories start at synapses where sights and sounds trigger responses, and end when genes are turned on in the nuclei of nerve cells to make permanent, physical changes in their connections.
When sensory information triggers known mechanisms near synapses, calcium is released into nerve cells, building up until it triggers chain reactions fine-tuned by partnering proteins like calmodulin or CaM, say the study authors. When calcium and CaM link up and arrive in a nerve cell’s nucleus, the compartment where genes operate, they set off reactions known to activate the protein CREB, which dials up the action of genes previously linked to memory formation.
Missing Link
Going into the study, a “missing link” in the field was an understanding of how synapses “talk to” nerve cell nuclei as memories form. In the current study, researchers determined for the first time that this communication occurs when gamma-CaMKII shuttles the calcium/calmodulin complexes that form just inside of nerve cells to their nuclei.
Comparing spatial memory in mice without gamma-CaMKII to normal mice, the study authors found that gamma-CaMKII “knockout” mice were much less able to locate a platform hidden beneath the surface of murky water in a maze. During this exercise, normal mice quickly identify the platform’s location.
The team also found that, an hour after maze training, normal mice displayed a significant increase in expression of three genes—BDNF, c-Fos, and Arc—known from past studies to help form long-term, spatial memories based on experiences. In contrast, training-induced increases in the expression of these genes did not occur in mice engineered to lack gamma-CaMKII.
Along with removing the entire gene encoding the gamma-CaMKII protein from some mice, a separate group of mice were engineered to have a version of the protein with a small change found by a 2012 study in a boy with severe intellectual disability. In the nerve cells of the boy, the protein building block at position 292 in the amino acid backbone of gamma-CaMKII, typically arginine, was occupied instead by a proline residue (R292P). The change rendered gamma-CaMKII a thousand times less able to trap the calcium-calmodulin complex, so it often arrived in nerve cell nuclei without its cargo.
Next steps for the team include determining how gamma-CaMKII fits into a larger “feedback machine” of nerve cell circuitry published by Dr. Tsien and colleagues in the journal Neuron in 2016.
“This learning machine, controlled by a key set of genes, senses nerve signaling levels and shapes sensory input into memories,” says Dr. Tsien. Experiments are planned to reveal more details about how the machine “copes with small flaws, including in those the gamma-CaMKII shuttle, but fails when too many problems build up in one or more of its components.”
Along with Dr. Tsien, NYU Langone authors were Samuel Cohen, Huan Ma, Benjamin Suutari, Nataniel Mandelberg, Natasha Tirko, Caitlin Mullins, Sandrine Sanchez, Ilona Kats, and Alejandro Salah, all within the Neuroscience Institute at NYU Langone and the Department of Neuroscience and Physiology. Dr. Tsien, Suutari, and Kats are also members of the Center for Neural Science at NYU. Making important contributions were authors Ma, Xingzhi He, Yang Wang, Guangjun Zhou, and Shuqi Wang in the Department of Physiology, Institute of Neuroscience, Zhejiang University School of Medicine, Hangzhou, China.
This work was supported by research grants from the National Institute of General Medical Science (GM058234); the National Institute of Drug Abuse (DA040484); the National Institute of Neurological Disorders and Stroke (NS24067); the National Institute of Mental Health (MH071739); the Druckenmiller, Simons, Mathers, and Burnett Family foundations; and a Medical Scientist Research Service Award (T32GM007308).
Media Inquiries
Greg Williams
Phone: 212-404-3533
gregory.williams@nyumc.org