


Women with mutations in the BRCA1 and BRCA2 genes are at dramatically higher risk of developing breast cancer.
ILLUSTRATION: STUART BRIERS
A woman’s lifetime risk of developing breast cancer in the U.S. is about 12 percent. But if she carries inherited mutations in the BRCA1 and BRCA2 genes, which account for 5 to 10 percent of all breast cancer cases, her risk is dramatically higher—anywhere from 40 to 90 percent, depending on individual risk factors.
In a recent study published in Nature, and funded partly by the Breast Cancer Alliance, researchers at NYU Langone describe a new way to potentially slow or halt the growth of cancers linked to BRCA1 and BRCA2 mutations. In healthy cells, the BRCA genes encode proteins that reliably repair damaged DNA. In mouse and human cells with BRCA1 or BRCA2 mutations, however, the researchers discovered that a backup repair strategy takes over. This much less reliable mending kit employs an error-prone enzyme called polymerase theta, encoded by the POLQ gene, which may fuel cancerous growth.
“One way to kill cells with BRCA mutations is to block this enzymatic pathway and deplete POLQ,” says NYU Langone researcher Agnel Sfeir, PhD, a member of the Laura and Isaac Perlmutter Cancer Center.
But a cancerous cell is still a living cell. From the cell’s vantage, this backup polymerase strategy is potentially lifesaving as it helps stave off full-blown genetic havoc. Agnel Sfeir, PhD, an investigator at the Skirball Institute of Biomolecular Medicine and a member of the Laura and Isaac Perlmutter Cancer Center, says this observation suggests an entirely new way to target BRCA cancers. “One way to kill cells with BRCA mutations is to block this enzymatic pathway and deplete POLQ,” she says.
The finding stems from what Dr. Sfeir describes as a “simple” question about basic biology. The tips of our chromosomes, called telomeres, contain multiple repeats of the DNA letters TTAGGG and are normally protected by protein caps that act like the plastic nibs on shoelaces. If left unprotected, however, the telomeres can decay or fuse together, sometimes leading to massive DNA rearrangements like those seen in cancer. Which DNA letter, Dr. Sfeir wondered, appears at the precise junction between two abnormally fused telomeres? “You answer the question, and you don’t know where the science will take you,” she says.
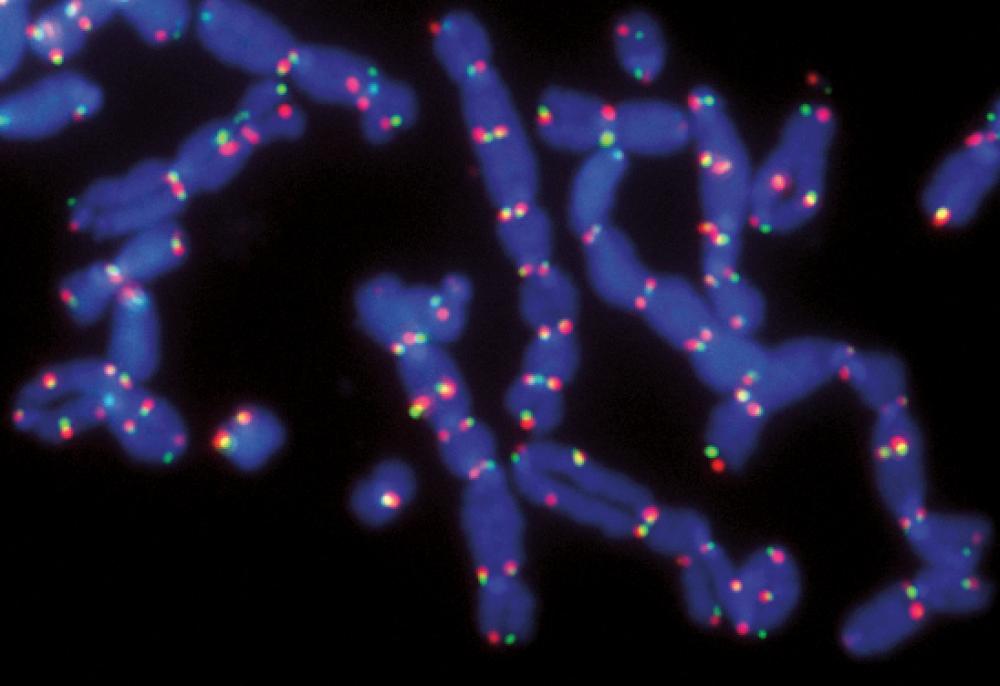
The tips, or telomeres, of these mouse chromosomes have become dysfunctional, causing individual chromosomes to fuse together in long tangled strands of DNA.
Image: Agnel Sfeir
When the researchers sequenced the DNA at multiple telomere fusions, they unexpectedly found that many of the junctions contained randomly inserted DNA letters instead of the TTAGGG repeats. The random insertions suggested the activity of an error-prone polymerase. After testing several kinds of polymerase, the collaborators found that knocking out the POLQ gene for polymerase theta significantly reduced the propensity of dysfunctional telomeres to stick together, suggesting that the enzyme might actually promote the phenomenon. In particular, the data suggested that this polymerase plays a key role in the highly unreliable DNA patching service that may trigger both chromosomal fusions and wholesale rearrangements in cells with BRCA mutations.
Together with collaborators from The Scripps Research Institute in La Jolla, California, and the University of Texas at Austin, Dr. Sfeir’s lab found that blocking the activity of POLQ in cells lacking the BRCA genes, meaning those deprived of both DNA repair mechanisms, led to chromosomal chaos and markedly reduced survival. Her lab is now testing whether the same POLQ-targeting strategy can inhibit tumor formation in mouse models of breast cancer.
A simple question and observation, Dr. Sfeir says, has since opened the door to an entirely new line of scientific inquiry. “There are many types of cancers that have altered DNA repair, and this polymerase theta seems to be up-regulated in many tumor cells,” Dr. Sfeir says. “So why? What is it doing? Why is it up-regulated? All of these are important questions. We’re going to be busy for the next 10 years or so.”

